I was appointed associated professor in 2017 at Université Paris Diderot in the CMPLI laboratory. Since my appointment, I have initiated a research project focusing on predicting the structures of protein-peptide complexes using my solid skills in in silico approaches, and in particular the simulation of protein-ligand interactions, data analysis and modelling.
Since 2020-2021 and the publication of the Alphafold 2 programme, structural bioinformatics approaches have been transformed. A major part of my research has involved integrating this revolution into my research, and several research projects have had to be radically reoriented. Alphafold and its derivatives have been integrated into the RPBS platform on a regular basis, both technically (software installation) and in terms of calls for tenders (AAP Idex ‘Gros Equipements’ GPU-APB, 144 k€, 2022) or services and collaboration (projects underway with a team from the Gustave Roussy Institute and a team from I-STEM).
These collaborations with experimental teams are particularly important to me. In the course of my research, 21 of the 30 articles published in peer-reviewed journals (including JACS, Science Advances, Structure, EMBO J, 3 PNAS and 4 NAR) have been produced in collaboration with experimentalists; for 18 of these articles I am the main author of the simulation/modelling part and corresponding author for 3 publications.Open Source software
Since joining CMPLI and RPBS, I have developed several open-source python libraries to make Gromacs molecular dynamics simulation tools and several molecular docking tools such as Autodock and Vina more accessible to non-specialists.
I also developed a python library to manipulate coordinate files and analyze AlphaFold results.

Web services
Online web service Since my arrival on the RPBS platform, I have become familiar with putting web services online. In 2021 I developed the SeamDock online service (deployed on the RPBS platform : https://bioserv.rpbs.univ-paris-diderot.fr/services/SeamDock/), which integrates various molecular docking tools into a common framework and enables global and/or local docking of ligands to a receptor. This tool can be used in collaborative mode with partners from all over the world or in a classroom focusing on docking methods or receptor-ligand interactions, while ignoring software installation and Unix command lines.
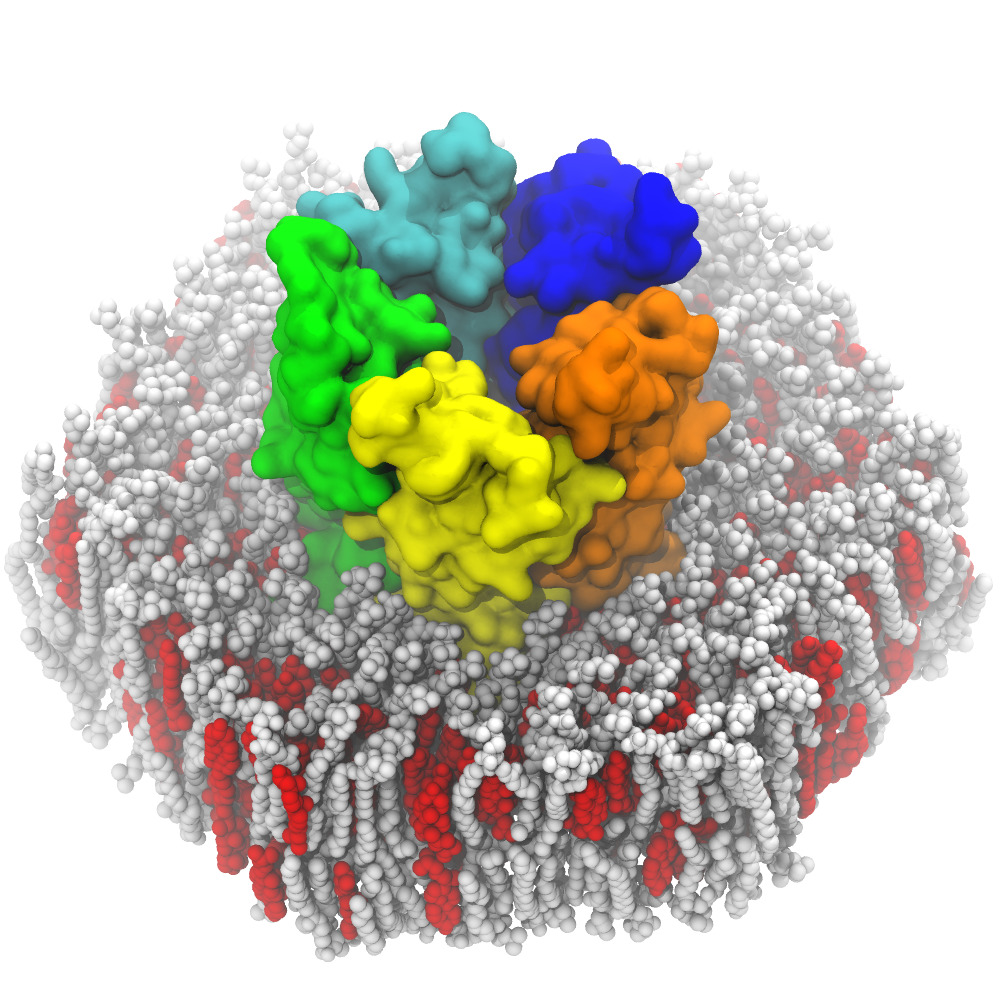
Protein-Peptide Interactions
Proteins play a key role in almost all cellular functions. These macromolecules rarely act in isolation, but within complex interaction networks. Recent methods of characterizing protein-protein interactions (PPI) have made it possible to obtain high-throughput data on these interaction networks. These protein-protein interactions act usually via large interfaces (1500-3000 Å2) with no well-defined binding pocket.
The modulation of these PPIs has many therapeutic applications, but the physical characteristics of these interfaces make the rational design of inhibitors difficult. The use of peptide ligands mimicking a portion of the interaction surfaces overcomes these difficulties. These molecules are also used in a natural way, indeed peptide-protein interactions represent 15 to 40% of all cellular interactions.
These polymers have a high potential for therapeutic applications, so it is not surprising that many numerical tools have been developed to predict the structure of peptide-protein complexes.
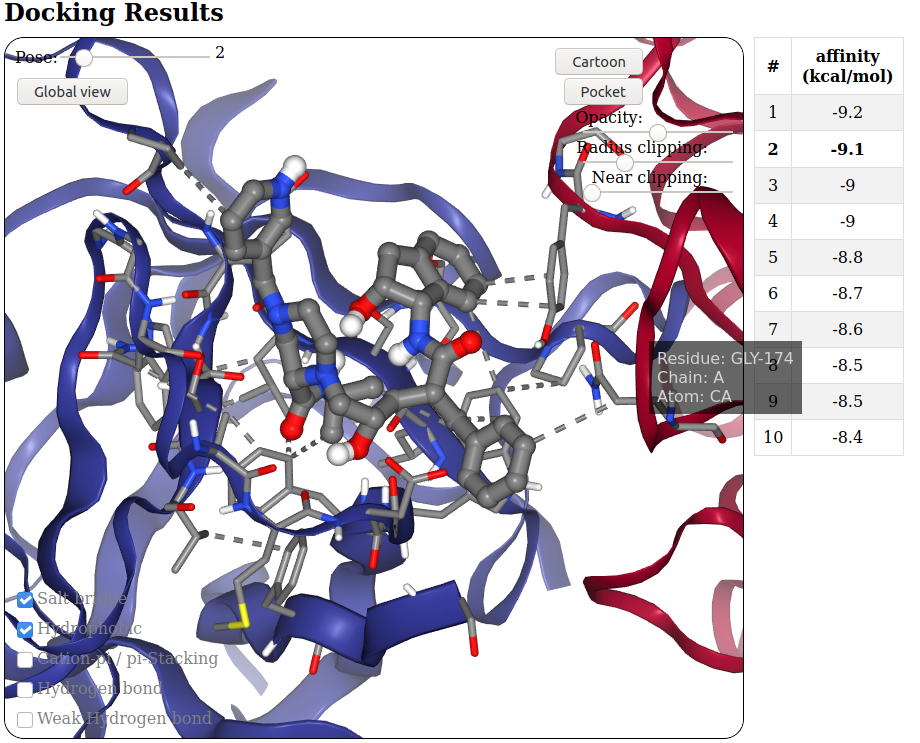
To tackle the challenges of peptide-protein interactions we use extensive molecular dynamic (MD) simulation of multiple peptide fragments with target proteins, and thus propose a breakthrough to overcome the major limitations of current approaches.
By using high precision physics-based approach, I intend to (i) identify peptide binding sites and especially key hotspots (ii) characterize hot spots peptide sequence specificity (iii) fold a target peptide in the best ranked binding sites and ultimately (iv) optimize the peptide sequence or predict de novo optimal peptide composition.
Gromacs_py
Gromacs_py is an open source python library allowing a simplified use of the gromacs MD simulation software. Gromacs_py can build topologie based on a pdb file, create the simulation system (box, add water and ions) and run minimisation, equilibration and production. One of the main objective of the gromacs_py wrapper is to automatize routine operations for MD simulation of multiple systems.
Synthetic Channels
Collaborations:- Mihail Barboiu European Institute of Membranes, Montpellier
- Marc Baaden IBPC, Paris
- Fabio Sterpone IBPC, Paris
A collaboration with a teams of chemists, the team of Mihail Barboiu, specialized in the design of materials mimicking the membrane functions (European Institute of Membranes, Montpellier). Inspired by the aquaporin and proton channel M2 of the influenza A virus, our collaborators synthesized a molecule to allow the selective transport of water molecules through the membrane. In the long term the aim of this project is to develop synthetic molecules for the purification and desalinization of water at lower cost.

Pentameric Channels
Collaborations:- Pluton Pullumbi Air Liquide, Jouy en Josas
- Marc Baaden IBPC, Paris
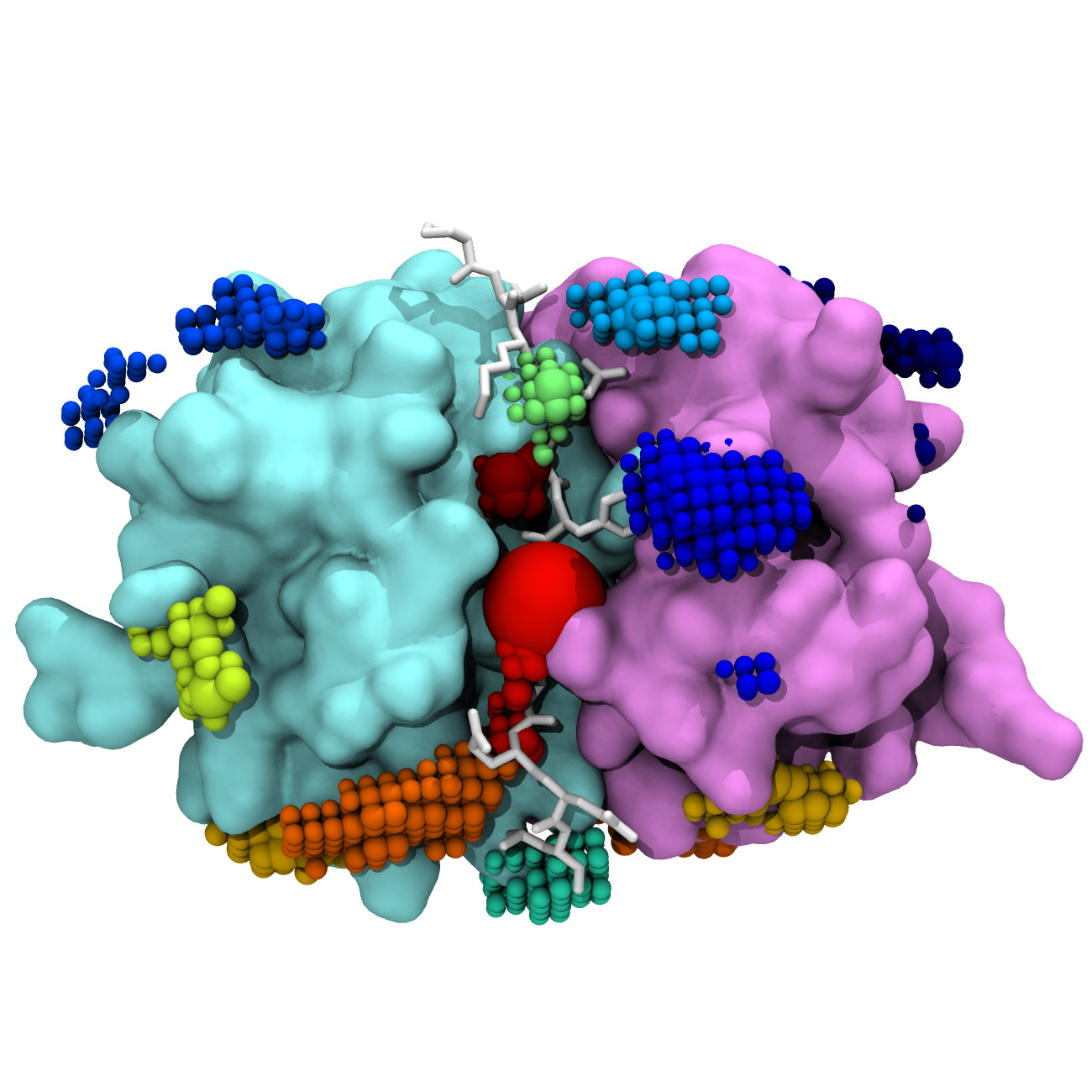
Pentameric ligand-gated ion channels (pLGIC) are essential components of cell-cell communications in the nervous system; their channel opens upon the binding of a neurotransmitter (gating mechanism) and lets ions flow through the pore down the electrochemical gradient (permeation). Understanding this transition is crucial as potentiating molecules such as anesthetics and recreational drugs are believed to affect transition barriers or the relative free energy of states.
This work focuses on GLIC, a prokaryotic homologue, the structure of which has been solved in three distinct functional states. Using molecular dynamics (MD) simulations reaching an aggregated time of 100 microseconds, we aim to characterize the transition between the different states, and how it can be modulated.